Das Megazystis-Mikrokolon-intestinale Hypoperistaltik-Syndrom (MMIHS) ist eine sehr seltene angeborene Erkrankung mit den namensgebenden Hauptmerkmalen zu große Harnblase (Megazystis), Mikrokolon, verminderte bis fehlende Peristaltik (Hypoperistaltik) des Darmes. S. 461
Das Syndrom wird von der Datenbank OMIM in Familiäre viszerale Myopathie eingegliedert.
Synonym: Berdon-Syndrom
Die Namensbezeichnung bezieht sich auf den Erstautor der Erstbeschreibung aus dem Jahre 1976 durch den US-amerikanischen Kinderradiologen W. E. Berdon und Mitarbeiter.
Verbreitung
Die Häufigkeit ist nicht bekannt, bislang wurde über 230 Betroffene berichtet, davon waren 71 % weiblich. Die Vererbung erfolgt autosomal-dominant oder autosomal-rezessiv.
Ursache
Die Ursache ist nicht bekannt. Es werden genetische, neurogene, myogene und hormonelle Faktoren diskutiert.
Der Erkrankung können folgende Mutationen zugrunde liegen:
- im ACTG2-Gen auf Chromosom 2 Genort p13.1, 44 %, autosomal-dominant
- im LMOD1-, MYH11-, MYL9- oder MYLK-Gen, jeweils Einzelfälle, autosomal-rezessiv
in etwa 55 % ist die Ursache unbekannt.
Klinische Erscheinungen
Klinische Kriterien sind:
Die Neugeborenen fallen durch vorgewölbtes Abdomen aufgrund der massiv vergrößerten Harnblase ohne Spontanentleerung auf. Hinzu können galliges Erbrechen und ausbleibender Mekoniumabgang kommen.
- Megazystis ohne spontane Entleerung, Dauerkatheter erforderlich
- Hydroureteren, Hydronephrose, eventuell Bild des Prune-Belly-Syndroms, aber ohne Abflusshindernis
- Mikrokolon mit ausgedünnter Muskelschicht bei normalen Ganglienzellen, eventuell verkürztes Mesenterium, dilatierter Dünndarm und/oder Malrotation
- Hypoperistaltik, dauerhafte parenterale Ernährung notwendig machend
- Entwicklungsretardierung
- Gesichtsdysmorphien wie breite Stirn, Hypertelorismus, kurze Nase, kleine Mandibula, eventuell Gaumenspalte, Ohrmuscheldysplasie
Hinzu können Herzfehler, Klumpfuß, Nagelhypoplasien kommen.
Diagnose
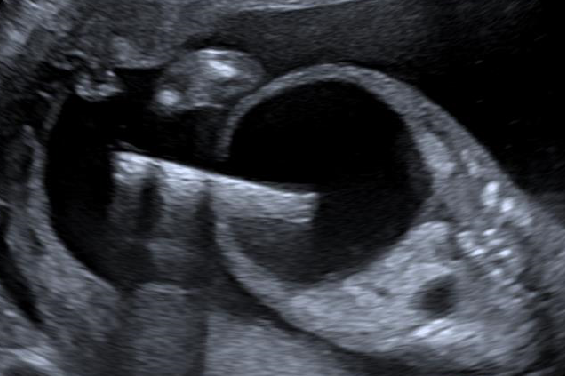
Das Leitsymptom der vergrößerten Harnblase mit Auftreibung des Bauches kann schon pränatal durch Sonografie und Feinultraschall ab dem 2., das begleitende Polyhydramnion ab dem 3. Trimenon erkannt werden. Der Einsatz der Magnetresonanztomographie ist gleichfalls möglich.
Die Diagnose stützt sich nach der Geburt auf die Klinik sowie bildgebende Verfahren wie Sonografie, Röntgenaufnahme und Kolonkontrasteinlauf.
Differentialdiagnose
Abzugrenzen sind:
- Chronische intestinale Pseudoobstruktion (CIPO)
- Harnwegsobstruktion
- Kongenitales Megakolon
- Darmatresien
- Analatresie (anorektale Malformation)
- Mekoniumileus, Mekoniumpfropfsyndrom
- Hypothyreose
- Neugeborenensepsis
- medikamentös z. B. Magnesiumsulfat, Opioide
- Embryofetopathia diabetica
- Prune-Belly-Syndrom
- Multisystemische Dysfunktion der glatten Muskeln (MSMDS)
Therapie
Eine durchgreifende Behandlung gibt es bislang nicht. Meist wird eine totale parenterale Ernährung erforderlich. An eine Multi-Organtransplantation kann gedacht werden. Die Lebenserwartung gilt als kurz, meist aufgrund von Sepsis oder Multiorganversagen.
Literatur
- P. Puri: Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome. In: A. Holschneider, P. Puri (Hrsg.): Hirschsprung’s Disease and Allied Disorders. Springer, Berlin / Heidelberg 2008, S. 267–273, doi:10.1007/978-3-540-33935-9_19, Print ISBN 978-3-540-33934-2, Online ISBN 978-3-540-33935-9
Weblinks
- MMIHS
- Rare Diseases
Einzelnachweise